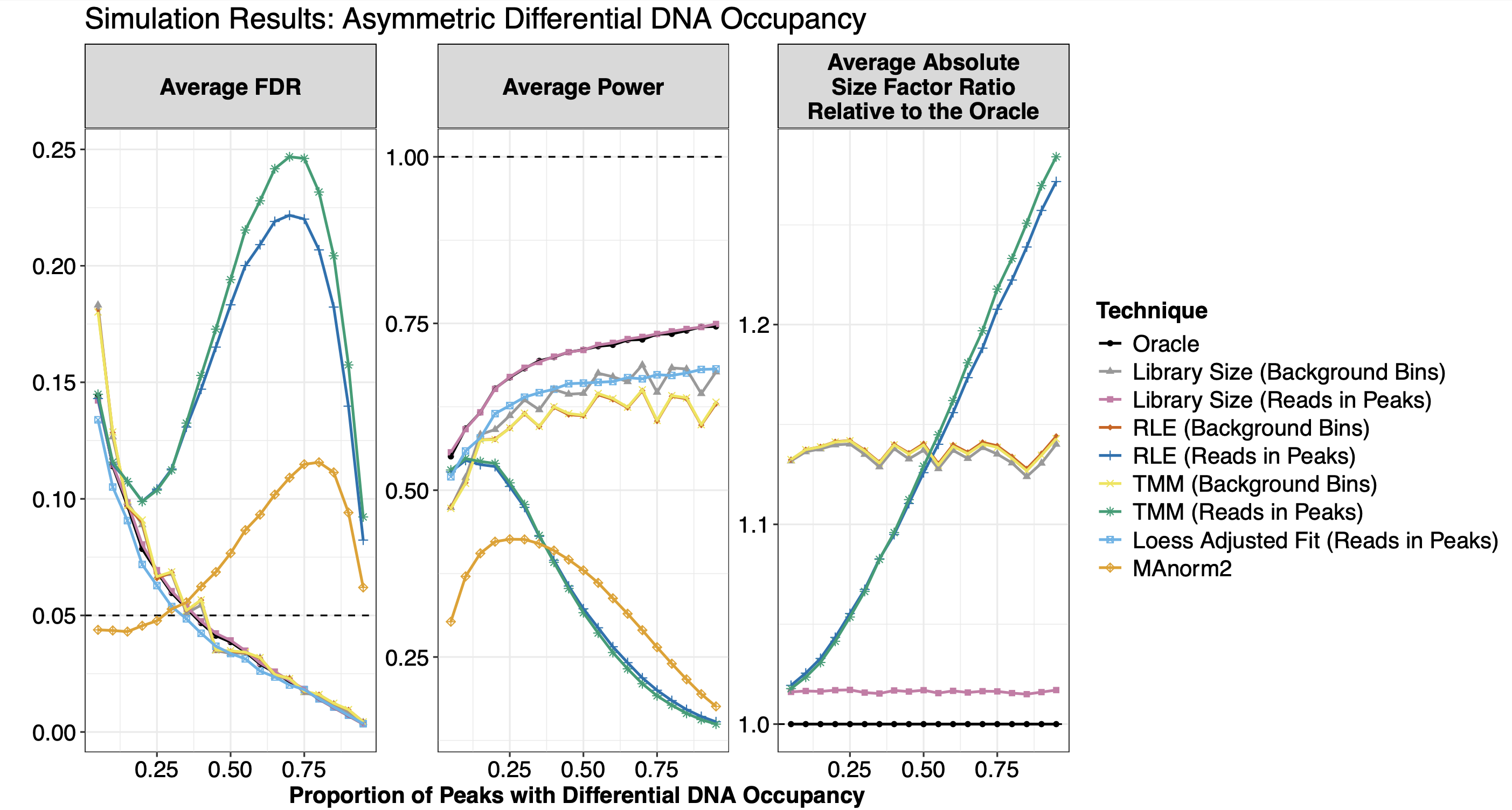
Overview: Chromatin immunoprecipitation with high-throughput sequencing (ChIP-seq) provides insights into both the genomic location occupied by the protein of interest and the difference in DNA occupancy between experimental states. Given that ChIP-seq data are collected experimentally, an important step for determining regions with differential DNA occupancy between states is between-sample normalization. While between-sample normalization is crucial for downstream differential binding analysis, the technical conditions underlying between-sample normalization methods have yet to be examined for ChIP-seq. We identify three important technical conditions underlying ChIP-seq between-sample normalization methods: balanced differential DNA occupancy, equal total DNA occupancy, and equal background binding across states. To illustrate the importance of satisfying the selected normalization method’s technical conditions for downstream differential binding analysis, we simulate ChIP-seq read count data where different combinations of the technical conditions are violated. We then externally verify our simulation results using experimental data. Based on our findings, we suggest that researchers use their understanding of the ChIP-seq experiment at hand to guide their choice of between-sample normalization method. Alternatively, researchers can use a high-confidence peakset, which is the intersection of the differentially bound peaksets obtained from using different between-sample normalization methods. In our two experimental analyses, roughly half of the called peaks were called as differentially bound for every normalization method. High-confidence peaks are less sensitive to one’s choice of between-sample normalization method, and thus could be a more robust basis for identifying genomic regions with differential DNA occupancy between experimental states when there is uncertainty about which technical conditions are satisfied.
Colando, S., Schulz, D., Hardin, J. (2025). Selecting ChIP-Seq Normalization Methods from the Perspective of their Technical Conditions, Briefings in Bioinformatics, 26(4). https://doi.org/10.1093/bib/bbaf43
Funded by Pomona College’s The Class of 1971 Summer Undergraduate Research Fund (Summer 2022) and the Kenneth Cooke Summer Research Fellowship (Summer 2024).